5 July 2020
New Research: Sensing sugar

In this post, I would like to discuss an research paper that made some surprising discovery about how we sense sugar. The article starts out with the astonishing fact that the average American consumes more than 45kg of sugar per year. The paper then investigates how we sense sugar. And equally astonishingly, it is not (only) via the mouth.
According to common knowledge, sweet compounds are detected by taste receptors on the tongue and palate epithelium. These taste receptors also sense artificial sweeteners, which were introduced more than four decades ago. However, when given to mice, artificial sweeteners are not as rewarding as real sugars in long-term choice tests.
This suggested that perhaps something is different about sensing of real sugar and artificial sweeteners. Indeed, it has been previously observed that mice lacking the ability to taste sweet compounds still develop a preference for sugar. And this sweet taste receptor-independent craving for sweet food is only elicited by real sugar, but not by artificial sweeteners.
The paper began with an interesting experiment, in which the researchers exposed mice to either sugar solution or artificial sweetener (acesulfame K) solution. Initially, the mice showed no preference. However, after half a day the mice started to switch to the sugar solution, eventually almost exclusively drinking the water containing glucose. The same response also happened in mice that lacked sweet receptors on their tongue and palate. These results hence suggested that sugar, but not artificial sweetener, can be sensed after ingestion via the mouth.
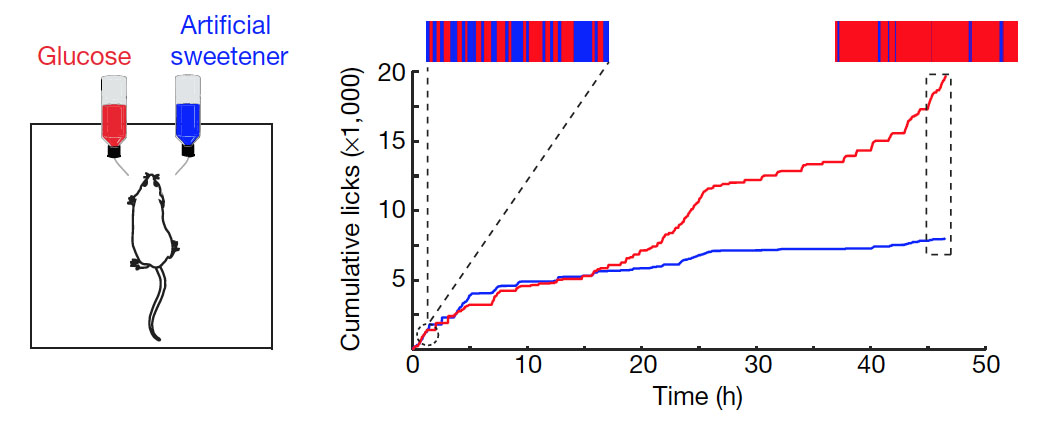
Sugar activates a gut–brain sugar sensing axis. Left figure: Mice were allowed to choose between 600 mM glucose and 30 mM acesulfame K artificial sweetener. Preference was tracked by electronic lick counters, and the experimental results are presented in the right figure: The bars on top of the figure illustrate the lick frequency for glucose (red) versus artificial sweetener (blue) during the first and last 2,000 licks of the behavioural test. The results show that initially the mice have no preference, but by 48 h a clear preference for glucose over artificial sweeteners can be observed.
The researchers hypothesized that sugar may be detected by specific sensory neurons in the gut and stimulate particular brain regions that mediate sugar preference. Thus, the researchers tried to identify neurons that become activated when giving mice glucose. They did this by staining sections of the brain to detect increased expression of the transcription factor c-fos, a marker of neuronal activation. They found that a region called the caudal nucleus of the solitary tract (cNST) becomes activated upon ingestion of sugar, but not artificial sweetener or water. Importantly, the cNST brain region also became activated when the sugar (but not sweetener) solution was infused directly into the stomach, bypassing taste receptors in the mouth.
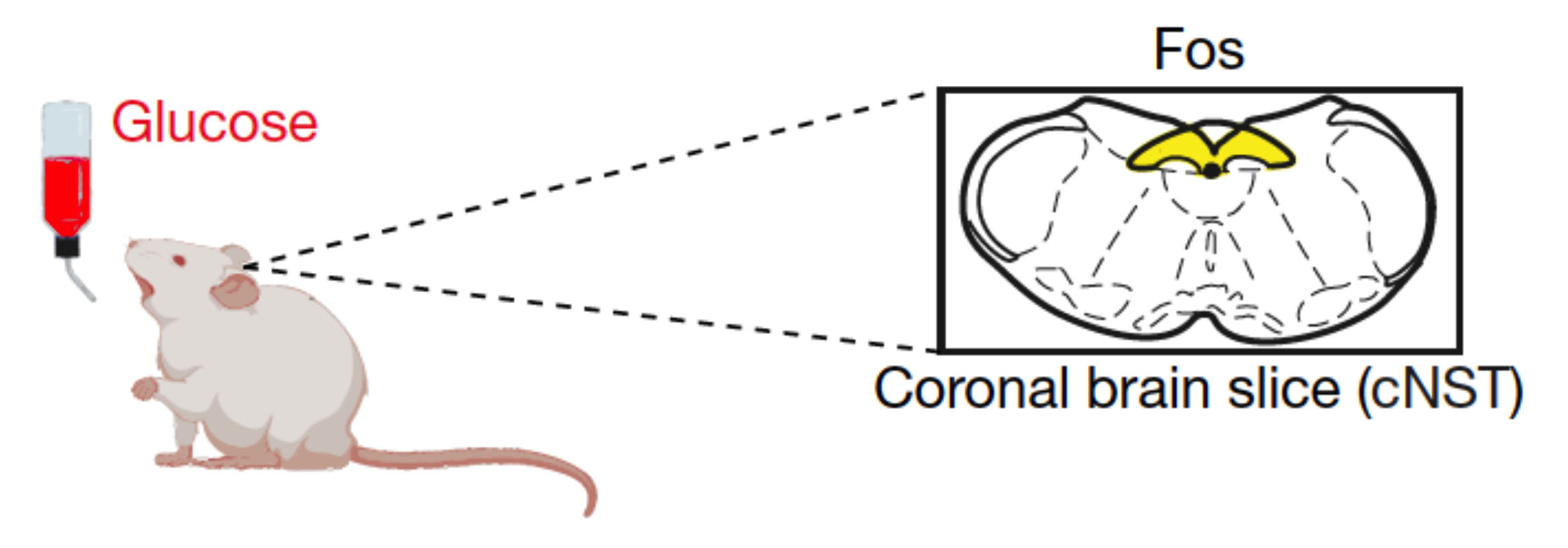
Detection of glucose in the gut leads to the activation of the caudal nucleus of the solitary tract (cNST) in the brain.
The researchers then hypothesized that the signal from the gut to the brain is transduced via the vagal nerve. To prove this, they performed a rather complicated experiment. They monitored the activation of the cNST region in the brain via a techniques called fibre photometry. They expressed in the cNST neurons a genetically encoded fluorescent sensor protein that becomes fluorescent when the calcium concentration in the neuron increases. An increase in the cellular calcium concentration indicates activation of the neuron. To detect the increased fluorescence, they implanted an optical fibre into the brain near the cNST region. When they then delivered glucose into the intestine (duodenum) via a catheter, they observed that the fluorescence in the excitatory cNST neurons increased. Importantly, this response was absent in mice with disrupted vagal nerves. This confirmed the presence of a gut-brain sugar sensing axis that is mediated by the vagus nerve.
In another complex experiment, the researchers showed that the neurons in the cNST region of the brain are required for the sugar preference. Showing that something is required usually involves removing it and seeing if the effect is still there. Hence, to determine if the neurons are required for sugar preference, the researchers removed these neurons and determined if the mice loose the preference for sugar. To specifically remove the cNST neurons, the authors expressed the tetanus toxin in these cells, which leads to cell death. The DNA encoding the tetanus toxin was delivered into the cNST region via Adeno-associated virus (AAV), which was injected into the specific brain region. However, expression of the tetanus toxin required recombination of the coding DNA sequence, which was mediated by a Cre recombinase. The Cre recombinase coding sequence was placed downstream of a promoter that becomes activated by c-fos (a transcription factors that become activated upon neuronal excitation). Hence, in the actual experiment, the virus was injected into the brain region, and then the mice were given sugar water. This led to the activation of the cNST neurons, activation of the c-fos transcription factor, expression of Cre recombinase, recombination of the tetanus toxin coding sequence and subsequent expression of tetanus toxin in these neurons. This resulted in death of the sugar activated neurons.
When subsequently the researchers gave the mice glucose again, a preference for glucose was no longer observed, showing that the cNST neurons are indeed necessary for sugar sensing in the gut – a rather complicated experiment to show that activation of cNST neurons is required for sugar preference. But nonetheless an important one, which may in the future even have therapeutic implications, because it suggests potential therapeutic targets to inhibit sugar craving in humans.
With all this knowledge, the researchers then tried to rewire the whole pathway and make mice crave a different stimulus, much in the spirit of physicist Richard Feynman, who said, “What I cannot create, I do not understand”. To do this, they first identified the exact neurons in the cNST brain region that mediate the sugar preference. This was accomplished by stimulating mice with sugar and then staining sections of the cNST region for the transcription factor c-fos (to label the neurons that became activated). The researchers found that the activated neurons are positive for expression of the specific neuronal marker protein proenkephalin. Hence, sugar activates specific neurons that express proenkephalin. The authors then expressed a synthetic “designer” receptor in these proenkephalin-positive neurons. The designer receptor responds to the drug clozapine, which binds to the receptor ligand binding domain on the cell surface. Binding of clozapine to this designer receptor then results in the activation of the neurons.
In the actual experiment, the researchers gave mice artificially sweetened cherry-flavoured and grape-flavoured solutions as drinking water. The grape solution was mush less sweet, but contained clozapine. What the researchers found was that after 48h the mice switched to the less sweet, but clozapine containing grape-flavored solution. This suggests that the clozapine, after ingestion and travel through the blood circulation, was able to activate the designer receptor expressing cNST neurons and mediate preference for grape-flavored solution. This confirms that activation of the specific cNST neurons was enough to create sugar preference. And the mice expressing the synthetic receptor for clozapine could in theory made to eat anything, as long as clozapine was added to the food.
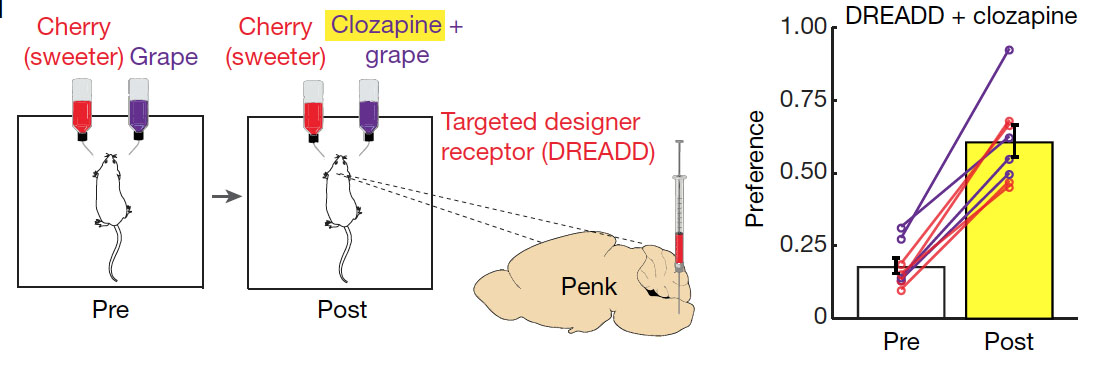
In the experiment, a plasmid encoding for the clozapine designer receptor (called DREADD) was injected into the cNST area. The designer receptor was only expressed by glucose sensing proenkephalin neurons, which was achieved by using a similar approach as described above for the tetanus toxin (i.e., expression of the DREADD receptor required a recombination event, which was mediated by Cre-recombinase, which in turn was expressed from the proenkephalin gene promoter). Upon expression of the clozapine receptor in proenkephalin positive cells, the mice were then tested for their preference between two flavours for 48 h. Shown is an example using cherry-flavoured drink (containing 2 mM acesulfame K artificial sweetener) versus grape-flavoured drink( with 1 mM acesulfame K). After initially conditioning the mice (Pre), clozapine was added to the less-preferred flavour. This resulted in a switch in preference to the less sweet grape solution, which is shown in the diagram on the right. The preference for the less sweet grape solution was initially less than 0.5 and hence not preferred. After adding clozapine to the less sweet grape solution, the preference for this increased in all mice to above 0.5 (which means the mice licked the less sweet grape solution more often than the sweeter one).
The study uncovered the entire neuronal pathway through which sugar preference is mediated. But it did not really clarify how the gut senses the glucose taken in by the mice. There are different ways through which the gut can sense external stimuli. There could be peripheral sensory cells in the gut mucosa. Alternatively, the sensory cells could be located further up and send sensory nerve endings to the stimulant site to detect specific external signals (see the diagram below taken from wikipedia).
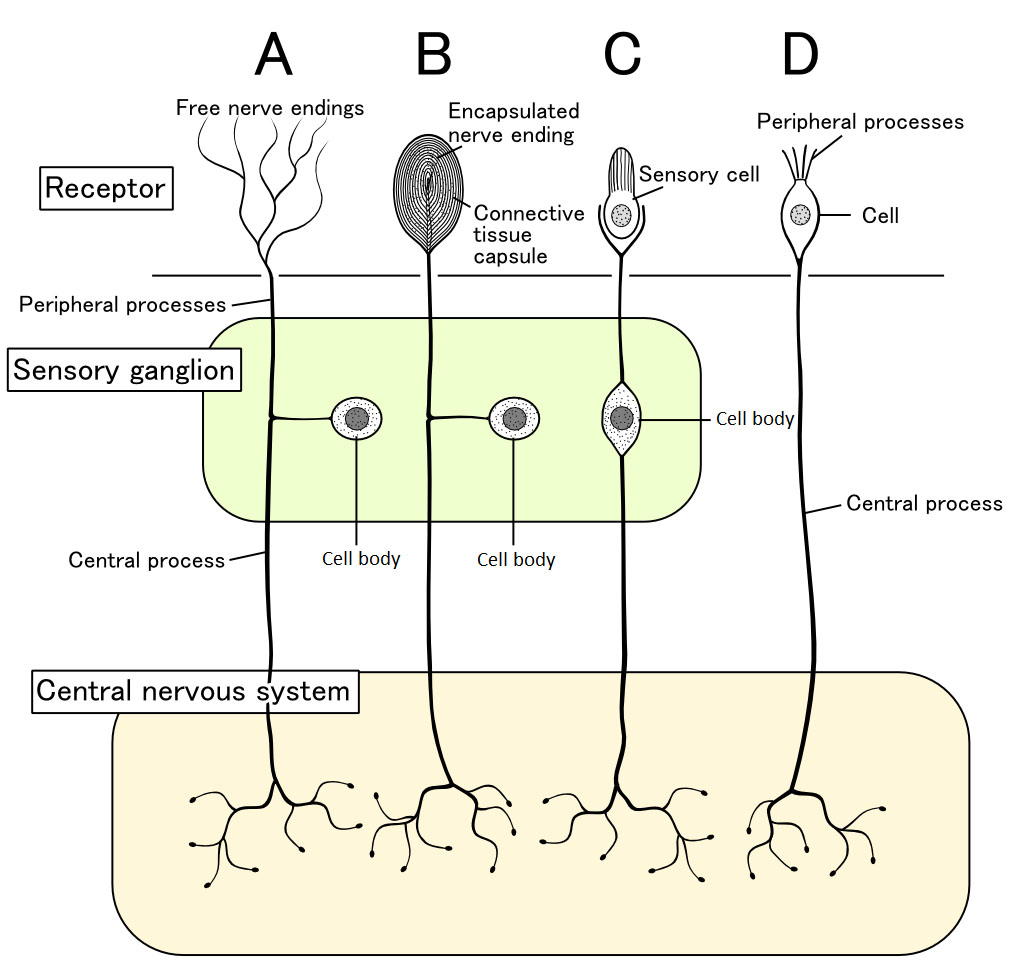
In the case of glucose sensing in the gut, it seems likely that there must be sensory neurons in the mucosal wall of the intestine, which upon stimulation by glucose activate vagal nerve endings in the gut. As a first step to characterize the sensing mechanism, the authors have shown that glucose sensing in the gut is dependent on the main glucose transporter in our gut, SGLT1. Interestingly, the glucose analogue 3-O-methylglucose, which can also be taken up via SGLT1 but is not further metabolized in cells, can also stimulate the gut-brain sugar axis. This suggests that glucose metabolism is not necessary for the sensing mechanism. Also of note, fructose is not a substrate for SGLT1, and as expected, fructose does not activate this sugar sensing pathway. This is likely a reason why the combination of glucose and fructose (in table sugar or high-fructose corn syrup) is particularly harmful: Glucose mediates the craving and fructose, taken up by the gut by other sugar transporters, mediates the adverse effects via its metabolism in the liver.
In conclusion, the study has described a novel mechanism through which glucose can be sensed in our gut. This may have important implications. For instance, the study identified a number of potential targets in the gut-brain sugar sensing axis to develop drugs to inhibit sugar craving. The authors also suggest that “it may be possible to develop a new class of sweeteners that activate both the sweet-taste receptor in the tongue and the gut–brain axis, and consequently help to moderate the strong drive to consume sugar.”